生物制品质量控制 
分析方法转移、验证/确认(理化、生化、细胞及微生物);
中间体、原液及制剂成品的放行检测;
公用系统(水、环境、气体等)的取样与检测;
原辅料及包装材料的放行检测。
 EN
EN
EN


分析方法转移、验证/确认(理化、生化、细胞及微生物);
中间体、原液及制剂成品的放行检测;
公用系统(水、环境、气体等)的取样与检测;
原辅料及包装材料的放行检测。

BSL-2 认证,可独立承担 AAV、细胞产品、活菌微生态产品及其他 BSL-2 等级样品的检测与方法验证服务;
消毒剂验证服务。

稳定性研究方案设计;
稳定性样品的储存、取样与检测;
稳定性研究趋势分析;
稳定性研究总结与可比性分析
|
板块 |
服务项目 |
主要职责与能力 |
|
原辅料控制 |
对所有进厂物料(包括培养基、试剂、辅料及内包装材料)进行检测与放行。依据 USP/EP/ChP 标准建立质量标准,执行鉴别、效价及安全性检测,从生产源头确保合规。 |
依据 USP/EP/ChP 标准建立质量标准,执行鉴别、效价及安全性检测,从生产源头确保合规。 |
|
过程控制 |
在整个生产过程中实施过程控制(IPC)与过程检测(IPT)。实时监测滴度、浓度等关键属性,提供即时反馈,确保持续的过程受控。 |
实时监测滴度、浓度等关键属性,提供即时反馈,确保持续的过程受控。 |
|
放行检测 |
对原液(DS)和制剂(DP)执行全面的批放行检测。年检测能力超过 100 批次,已建立覆盖单抗、双特异性抗体、融合蛋白、疫苗等的平台。 |
年检测能力超过 100 批次,已建立覆盖单抗、双特异性抗体、融合蛋白、疫苗等产品的分析平台。 |
|
稳定性研究 |
设计并执行符合 GMP 和 ICH 指南的稳定性研究。配备受控稳定性试验箱,通过实时、加速及强制降解(强力破坏)研究评估产品降解途径与货架期。 |
配备受控稳定性试验箱,通过实时、加速及强制降解(强力破坏)研究评估产品降解途径与货架期。 |

|
模块 |
核心价值 |
主要服务内容 |
|
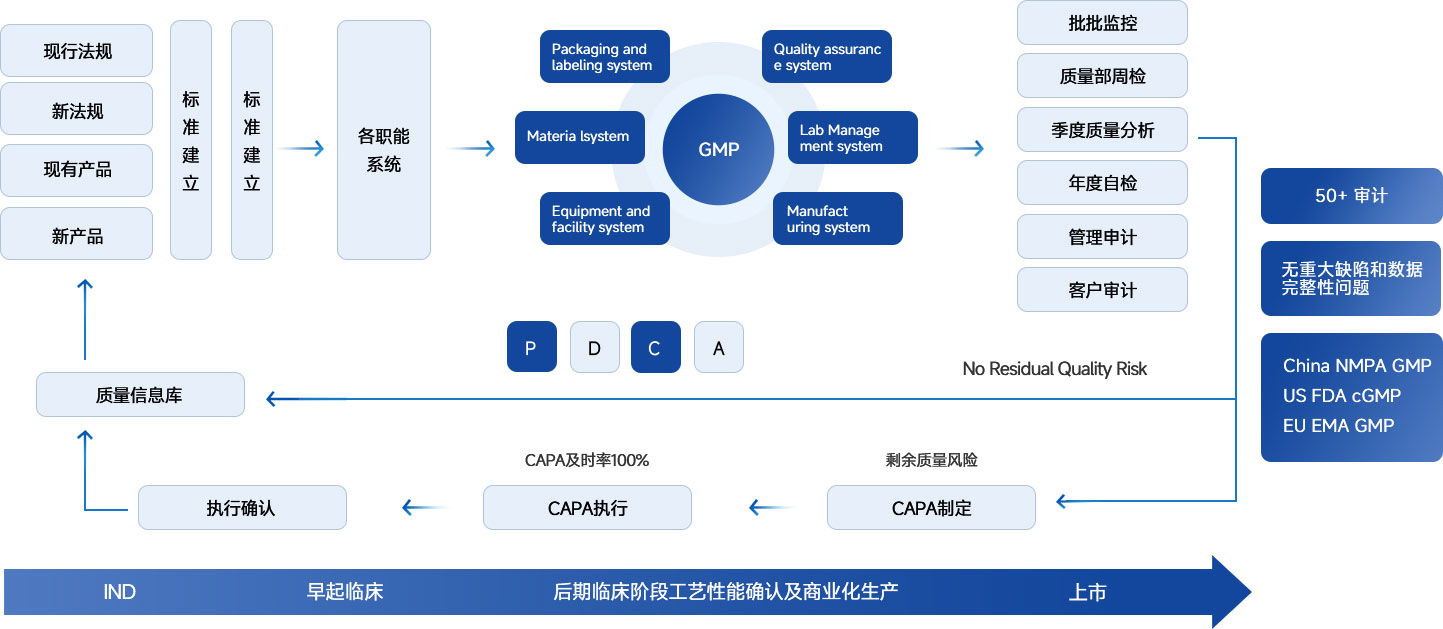
GMP 合规 |
确保运营完全符合全球药品生产质量管理规范(GMP)法规要求。 |
• 合规咨询:NMPA/FDA/EMA法规解读及策略建议。 • 体系建立:协助建立稳健的制药质量体系(PQS)。 • 新设施规划:为新设施/新产品制定合规计划。 • 审计服务:差距分析、模拟审计及供应商审计。 |
|
计算机化系统与数据 |
确保运营完全符合全球药品生产质量管理规范(GMP)法规要求。 |
• 体系建立:定义计算机化系统保障(CSA)框架。 • 合规支持:GxP评估、数据评估及控制策略。 • 验证与维护:系统验证执行及持续合规维护。 |
|
工程与设施设备 |
确保运营完全符合全球药品生产质量管理规范(GMP)法规要求。 |
• 工程管理:新设施规划、设计审核及良好工程规范(GEP)监督。 • 设备确认:从URS、FAT/SAT到3Q(IQ/OQ/PQ)的全生命周期指导。 • 法规对齐:跟踪并实施相关法规变更。 |
|
验证与风险分析 |
通过系统化的验证与风险管理确保持续的工艺稳健性。 |
• 验证管理:主导验证主计划(VMP)、工艺验证(PPQ)及清洁验证。 • 风险分析:包括清洁风险评估、交叉污染风险评估等服务。 |
|
MAH 服务 |
为上市许可持有人提供全链条质量运营支持。 • |
• 体系建立:协助搭建 MAH 质量体系。 • 运营指导:支持 B 证申请及日常质量运营。 • 审计管理:对合同生产组织及供应商进行审计。 |
|
CMC 服务 |
为药物开发和生产提供技术与注册支持。 |
• 技术转移:开展差距分析,审核转移方案及报告。 • 变更管理:支持场地转移及各类变更的注册申报。 • 工艺支持:为工艺表征、工艺验证及稳定性研究提供支持。 |
为药物研发提供全生命周期注册支持
支持向FDA/EMA/其他监管机构提交申报
准备Pre-IND会议/科学建议咨询申请
IND/CTA申报资料编制(CMC、非临床、临床)
IND/CTA提交及跟踪
临床开发阶段的变更管理
Pre-BLA申报资料及提交
BLA申报资料编制(CMC、非临床、临床)
批准后变更管理
批准后补充申请及生命周期管理(CMC)
支持上市后产品变更及补充/变更申请
批准后变更管理
变更实施(生产场地、生产工艺、处方、质量标准等)
变更风险评估
批准后补充申请资料的编制与提交

基于项目目标进行可行性评估与分析
制定策略、与监管机构的沟通计划及研发计划
编制项目启动与可行性报告
组织协调资料的汇编与审核
提供 CTD 模块的编制与审核,并完成 CMC 模块的填写与审核
协助准备问题答复、资料修订及补充
代表客户与监管机构沟通
为研发项目提供全流程支持,协助与监管机构沟通
制定注册策略及整个项目的详细实施计划
提供 CMC 策略及技术支持,定期审核研发进展
协助准备关键文件,如 CMC 开发计划、稳定性研究计划、杂质研究计划等
参与并支持关键研发里程碑及项目评审的准备工作
协助审评审批流程,跟踪审评进度及反馈
协助批准后变更管理及生命周期管理
协助 GMP 检查及 GMP 合规

业绩卓著:在中国、美国及欧洲拥有数十项成功的 IND/BLA 申报经验,覆盖双特异性抗体、重组蛋白等复杂分子。
顶尖团队:核心成员具备丰富的端到端注册事务经验,并拥有国际顾问网络支持,包括前 FDA 及 CDE 审评专家。
权威网络:与全球监管机构及行业协会保持长期密切合作,提供前沿政策洞察。

前瞻性策略:掌握不同国家的法规与审评理念,从早期研发阶段即可制定精准的注册与开发策略,提高成功率。
问题解决专长:擅长将技术证据与法规原理相结合,高效解决审评过程中的关键问题,加速审批进程。
标准化流程:建立了高效、标准化的文件编制、审核及提交流程,确保质量与时效性。
动态敏捷响应:实时跟踪法规更新,快速调整策略,确保申报始终符合最新标准,保持竞争优势。
在研发早期介入,基于全球趋势明确注册路径,并监督关键里程碑。
在研发早期介入,基于全球趋势明确注册路径,并监督关键里程碑。
代表客户进行高效、透明的监管沟通,目标不仅是获得批准,更是建立产品长期的市场竞争力。
